Auch seltene Kindererkrankungen mit schweren Schädigungen und funktionellen Beeinträchtigungen stehen in engem Zusammen-hang mit zugrundeliegenden zellulären Stoffwechselstörungen. Diese Stoffwechsel Prozesse helfen uns beim Verstehen der Krankheiten (bei deren Pathogenese). Und wir können auch schwere Erkrankungen über eine gezielte Stoffwechsel-Regulation und Ernährung Medizin therapeutisch beeinflussen!
Eine seltene Erkrankung ist der sogenannte Ornithintranscarbamylase-Mangel (OTC-Mangel; auch: Ornithintranscarbamylase-Defekt, OTCD). Und das Problem der Erkrankung ist, dass in Körper und in Gehirn der Kinder und Jugendlichen viel zu viel Ammoniak entsteht.
Ammoniak macht schläfrig und „dusselig“, Ammoniak verursacht Erbrechen, Atemstörungen, neurologische Symptome wie Ataxie, Tremor, Zittern, (das kann aussehen wie ein Morbus Parkinson), Nystagmus, Konzentrationsstörungen und eine hohe Infekt-anfälligkeit. Dies wird medizinisch häufig nicht richtig erkannt, denn die Beschwerden sehen aus wie ein Infekt und dann wie eine Absence.
Diese Erkrankung kann man behandeln, indem man Ammoniak bindet: Die Gabe des insulinunabhängigen Monosaccharids Galactose bindet Ammoniak und führt zur Synthese von Amino-säuren.
Zusätzlich gibt man höhere Gabe von Magnesium, Zink, niederdosiertes Kreatin; und man gibt morgens 100 Mikrogramm Selen.
Und man nimmt kurzkettige Kohlenhydrate, Pizza, Pasta, Cola weg! Typischerweise findet sich neben einem erhöhten Ammoniakspiegel auch erhöhter Harnstoff, erhöhte Harnsäure, erhöhte CK. Für diese Kinder sind Früchte wie Gift. Bereits ein Teelöffel Fruchtzucker erhöht den Blutdruck; das haben die betroffenen Kinder auch.
Wichtig sind eine fettreiche Ernährung und essenzielle Aminosäuren. Und man stärkt die Mitochondrien mit NADH, Coenzym Q10, Kreatin. Um abends um die Cortisolspiegel zu senken empfiehlt sich Ashwagandha und Melatonin.
Körperliche Regulation – Myoreflextherapie:
Neben diesem OTC-Mangel gibt es noch einige andere Erkrankungen, die mit dem erhöhten Ammoniak zu tun haben. Sogenannte Hyperammonämien.
Die Therapie ist jeweils ähnlich: Auch diesen Kindern kann man sehr gut helfen mit Glycoplan, sowie mit einer Darmbehandlung und Präbiotika.
Eine andere Hyperammonämie betrifft den Carnitin-Stoffwechsel. Bei Störungen des Carnitin-Stoffwechsels haben die Kinder und Jugendliche Probleme, dass Fett verbrannt wird, weil das Fett nicht in die Zelle kommt. Auch da helfen Stoffwechsel-Regulation und therapeutische Monosaccharide.
Einige Kinder haben auch Probleme mit dem Lysin-Stoffwechsel. Hier kann tatsächlich ein Substratmangel auch zu Hyperammon-ämie führen.
Es braucht also Labor-Messungen und eine gezielte Aminosäuren Therapie.
Nicht zuletzt gibt es auch Störungen im Energiestoffwechsel in den Mitochondrien. Je schlechter die Mitochondrien arbeiten (aufgrund von Insulinresistenzen), umso mehr entsteht Ammoniak.
Eine weitere Quelle von Ammoniak bei allen Hyperammonämien finden wir im Darm. Je höher der pH-Wert im Darm, umso höher der Ammoniakspiegel. Deshalb sollte der Darm jeden Tag mindestens einmal entleert werden. Hier hilft ggf. Magnesiumcitrat, Movicol und so weiter.
Phosphoglucomutase-1-Mangel (PGM-1-Mangel, PGM-1-Defizienz) und angeborene Defekte der Glykosylierung (CDG)
Ein PGM1-Mangel führt zu Glykosylierung Störungen: „Glykosylierung“ bezeichnet dabei die Anlagerung von Zucker-strukturen an Proteine oder Lipide. Dieser Vorgang ist für viele biologische Funktionen im Körper von großer Bedeutung. Wenn angeborene Störungen im Aufbau dieser Zuckerketten bzw. Zell-Antennen (Glykane) auftreten, kann dies beim Menschen zu schweren Erkrankungen führen, die häufig das Nervensystem betreffen. Diese Krankheitsgruppe wird als „Congenital Disorders of Glycosylation“ (CDG), also angeborene Störungen der Glykosylierung, bezeichnet.
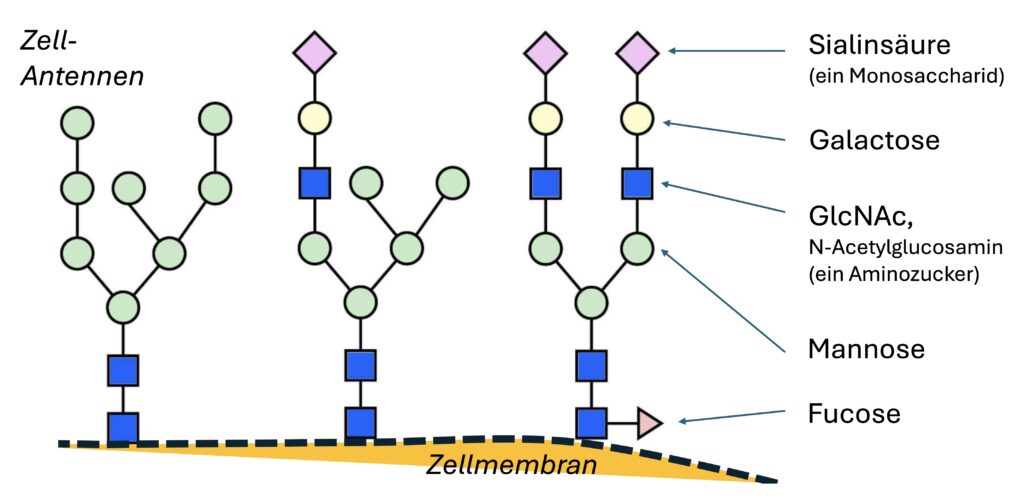
Die feinen Antennen der Zellen sind aus Zuckermolekülen aufgebaut. Und sie bestimmen so alle grundlegenden Prozesse im Körper: das Zellwachstum, die Zellreifung, die Entwicklung von Organen, die Signalübertragung zwischen Zellen sowie Immun- und Entzündungsreaktionen. Auch bei der Entstehung von Tumoren können sie beteiligt sein.
Die betroffenen Kinder erleben Entwicklungsverzögerungen; sie die haben erhöhte Leberwerte, Belastungen der Nieren, erhöhte Muskelenzyme und sie leiden unter Hypoglykämien (und können deshalb auch Anfälle erleben).
Kinder mit dieser Problematik profitieren erheblich von der Gabe von Galactose. Eineinhalb bis zwei Gramm pro Kilogramm Körpergewicht puffern die Hypoglykämien ab, führen zur Reparatur von der Zell-Antennen, regulieren das Gerinnungssystem, regulieren die Leberwerte und bringen die Niere wieder zurück in Funktion.
Ein Pionier im Zusammenhang mit genetischen Schwachstellen, Mitochondriopathien und diesem Defekt der Glykogenspeicher ist Prof. Dr. Thorsten Marquardt (Biochemiker und Arzt am Universitätsklinikum in Münster). Bei den betroffenen Kindern gelang es tatsächlich aufzuschlüsseln, dass diese Störungen in den Zell Antennen, reguliert werden. Der Verlauf und die Entwicklung der betroffenen Kinder zeigten, dass der ganze Stoffwechsel regulierbar war.
[ „Der Phosphoglucomutase-1-Mangel, der früher als Glykogenose identifiziert wurde, ist ebenfalls eine angeborene Glykosylierungs Störung. Die Gabe von Galaktose führt zu einer biochemischen Verbesserung der Glykosylierungsparameter in Zellen und Patienten, und die Zufuhr komplexer Kohlenhydrate stabilisiert den Blutzuckerspiegel.“
(Tegtmeyer, . . . Marquardt, T. (2014). Multiple phenotypes in phospho-glucomutase 1 deficiency. N Engl J Med, 370(6), 533-542. – Online: Pubmed ] [ > PGM1-Mangel: Milchzucker kann Leben retten. | > Vgl.]
Eine seltene Erkrankung ist die Adrenomyeloneuropathie (AMN; (eine Variante der Adrenoleukodystrophie, ALD). – Eine Erkrankung des Nervensystems und der Nebennieren. Im Verlauf der Erkrankung kommt es zu einer Demyelinisierung; also zur Schädigung der Myelinscheiden, der schützenden Isolationsschicht der Nervenfasern – insbesondere im Bereich des Rückenmarks.
Im krankheits-auslösenden Zusammenhang geht es um eine mitochondriale Dysfunktion, um zu viel oxidativen Stress, sowie um eine Störung der physiologische Fettverbrennung.
Wichtig für Hilfe und Entlastung ist, dass man ganz gezielt Proteine supplementiert, ferner alle Mineralstoffe und Vitamin D3/K2.
Konsequente neuromuskuläre Behandlungen / Myoreflextherapie sowie GALILEO Training helfen ebenfalls, den Krankheitsverlauf entscheidend zu lindern.
Die Metachromatische Leukodystrophie (MLD), eine neurode-generative Erkrankung im Kindesalter, führt normalerweise rasch in vier, fünf, sechs Jahren zum Tod.
MLD ist auch gekennzeichnet durch verdeckte Unterzuckerzustände. Das bedeutet: der Blutzucker erscheint normal, aber der HOMA-Index / also Insulin ist deutlich zu hoch. Diese Unterzuckerzustände schädigen das Gehirn und das Nervensystem.
Deshalb ist Ernährungsmedizin notwendig; also auch hier:
… Interdisziplinäre Zusammenarbeit bietet auch hier sehr weit-reichende Möglichkeiten!
Viele neuronale Entwicklungsstörungen hängen an veränderten Genen, und zwar im Bereich vom Calciumkanal, bei einigen im Bereich vom Kaliumkanal. So kommt es zu Störungen der Spannung in der Zellmembran, und auch zu Störungen im Einstrom von Calcium.
In der Tat haben wir schon vor ganz vielen Jahren festgestellt, dass Kinder mit Störungen von Kanaldefekten häufig einen erhöhten Homa Index HOMA-I haben, scheinbar normale Blutzucker, aber ein viel zu hohes Insulin. Deshalb leiden sie an Unterzuckerzuständen. Sie alle profitieren von Ribose, Galactose, Mannose.
Einzelne Kanaldefekte können über einen Bypass kompensatorisch behandelt werden. Beim Calciumkanaldefekt, beim CACNA1-Defekt, helfen hohe Dosierungen vom Zink, aber auch ordentliche Dosierungen vom Selen morgens bei 200 Mikrogramm. Alle betroffenen Kinder von Omega-3-Algenöl und auch von Proteins-hakes; insbesondere vom antientzündlichen Phytoprotect.
Das Dravet-Syndrom beginnt mit einer schweren, kaum zu behandelnden Epilepsie ganz früh. Die Kinder haben prinzipiell eine sehr schlechte Prognose. Die Kinder werden mit Antiepileptika behandelt und fallen häufig ins Koma.
Wir behandelten drei Kinder mit dem Dravet-Syndrom, die keine Anfälle mehr haben. Wir konnten dies Symptomatik regulieren über:
Fieberkrämpfe sind ebenfalls eine Folge der Hypoglykämie. Tatsächlich schaut man, dass eben zu viel Stress, zu viel Lautstärke, zu viel Aufregung, zu viel Licht, zu viel Computerarbeit und Handy vermieden werden sollte.
Tatsächlich behandeln wir (vor vielen Jahren schon) auch mehrere Kinder und Jugendliche, mit dieser Erkrankung. Spinozerebelläre Ataxien (spinocerebellar ataxias, SCA) bezeichnen eine Gruppe von neurodegenerativen Erkrankungen.
Koordinationsprobleme, Gleichgewichtsstörungen, Nystagmus, Tremor, Rigor, Störungen des Sprechens und der Feinmotorik.
Alle Kinder profitieren von:
Die Erkrankung der Mitochondrien steht schon seit 20 Jahren im Fokus unserer Arbeit. Deshalb untersuchten wir die Funktion der Mitochondrien bereits zu einer Zeit, in der das in Deutschland nicht üblich und kaum möglich war. Heute können wir die mitochondriale Funktion, die Dichte, die Qualität und die Verteilung von den Mitochondrien sehr elegant untersuchen: Der BHI, der bioener-getische Index war hier (Dank Burkhard Schütz und Brigitte König) ein Durchbruch in der Diagnostik.
Auch bei genetischen Schwachstellen kann man die Mitochondrien kompensatorisch behandeln. Die Schlüssel dafür:
In > Myoreflextherapie, Band 2 sind diese Erkrankungen näher erläutert.
Mitochondriopathien sind Störungen der zellulären Atmungskette; und sie können eigentlich in jedem Organ zuschlagen. Man nennt sie auch mitochondriale Zytopathie.
(Und in der Tat handelt sich auch um eine eigene Genetik der Mitochondrien, die weniger geschützt ist als die des Menschen.)
Bezeichnend sind:
…. Aber auch da hilft Ernährungstherapie. Und bei drei Kindern mit dieser Erkrankung waren wir in der Lage, mit fantastischen Eltern das Blatt zu wenden.
.. Diese neuromuskulären Störungen oder Syndrome korrelieren immer mit einer Nicht-Zöliakie-Glutenintoleranz. Also: Alle Mito-chondriopathien haben eine Sensitivität auf Gluten. Deshalb werden alle betroffenen Kinder glutenfrei und kuhmilch-frei ernährt. Häufig auch Nachtschattengewächs frei.
Die beiden mitochondrialen Multisystemerkrankungen, die meist im Kindes- oder Jugendalter beginnen, das MERRF-Syndrom und das MELAS-Syndrom, sind bei uns wenig bekannt.
Beim progressiv verlaufenden MERRF-Syndrom (Myoclonic Epilepsy with Ragged Red Fibers) stehen eher eine Ataxie und Myoklonien, Muskelzittern und Muskelschwäche, sowie Krampfanfälle im Vordergrund. Auch hier kann man früh über die Behandlung der Mitochondrien und des Darms regulativ Einfluß nehmen.
Beim MELAS-Syndrom (Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes) stehen eher eine kognitive Störung und Brain Fog, Aufmerksamkeitsstörungen, ADS, Chronic Fatigue und Kopfschmerzen im Vordergrund. Ganz vorne stehen auch dort wieder die Mitochondrien.
Das seltene MELAS-Syndrom zeigt sich meistens um das 8. Lebensjahr. Im Vordergrund stehen zunächst Kopfschmerzen, Migräne, Übelkeit und Erbrechen. Auch Sehstörungen und Paresen bei ungefähr 2/3 der Betroffenen und Hörverluste bei ¼ der Kinder sind typisch für diese mitochondriale Erkrankung. Am bedrohlichsten sind jedoch bei rund 90% der Kinder die epileptischen Anfälle.
Das Leigh-Syndrom ist eine seltene Erkrankung, die gewöhnlich im ersten Lebensjahr beginnt. In seltenen Fällen tritt sie auch bei Jugendlichen oder Erwachsenen auf.
Symptome im Säuglingsalter
Säuglinge mit Leigh-Syndrom können unter anderem folgende Symptome zeigen:
Zusätzlich können auftreten:
Im weiteren Verlauf der Erkrankung können sich die Symptome ausweiten auf:
Diese Stoffwechselentgleisungen können mit Schmerzen sowie Atem- und Nierenproblemen einhergehen.
Therapieansätze
In den offiziellen Leitlinien ist derzeit keine kausale Therapie für das Leigh-Syndrom beschrieben. Es ist jedoch bekannt, dass eine kleine Gruppe von Kindern mit einer speziellen Form des Leigh-Syndroms von Nahrungsergänzungsmitteln mit Thiamin (Vitamin B1) und Biotin profitieren kann.
Erfolgreiche Therapieansätze fokussieren sich auf:
Als Ernährungskonzept und Basisversorgung wird eine antient-zündliche Ernährung empfohlen:
Therapeutische Maßnahmen
Flankierend können unterstützend eingesetzt werden:
Ergänzende Maßnahmen (Übersicht)
Therapie & Anwendungen
Über die Zusammenarbeit mit der Charité Berlin, Professor Dieter Felsenberg und Professor Eckart Schöner der Kinderklinik in Köln und in Zusammenarbeit mit dem Dr. Tilman Görtler, schon 1990, sehen wir immer wieder Kinder mit Muskeldystrophie Beckerund Muskeldystrophie Duchenne.
Die Muskeldystrophie Duchenne beginnt meistens schon im ersten, zweiten Lebensjahr mit Entwicklungsverzögerungen, Schwierig-keiten beim Gehen, Springen, Treppensteigen, Müdigkeit bei einem Watschelgang und dann nimmt die Schwäche zu, Schwäche im Schultergürtel, Schwäche im Beckengürtel, dann kann das Herz betroffen sein.
Und in der Tat besteht hinter der Muskeldystrophie Becker, aber im Besonderen bei der Duchenne, eine verdeckte Insulinresistenz in der neuromuskulären Übergangszone. Deshalb: Ernährungs-medizin, Galileo-Training, massive Supplementationen, Behandlung der Darmgehirnachse und der Mitochondrien.
So kann man geistige Behinderungen und in vielen Fällen auch die Problematik vom Herz abfedern. In vielen Fällen kann man die Erkrankung, die sonst eigentlich spätestens im zwölften Lebensjahr im Rollstuhl endet, abfedern
Die Muskeldystrophie Becker bricht meist im 10.-12. Lebensjahr aus. Auch hier kann man mit Glycoplan, therapeutischen Mono-saccharide und mit ketogener Ernährung, mit Mitochondrien- und Stoffwechsel Medizin tatsächlich kompensatorisch aktiv sein (und tatsächlich Menschen behandeln, schon 1995, die auch heute noch ein normales Leben führen).
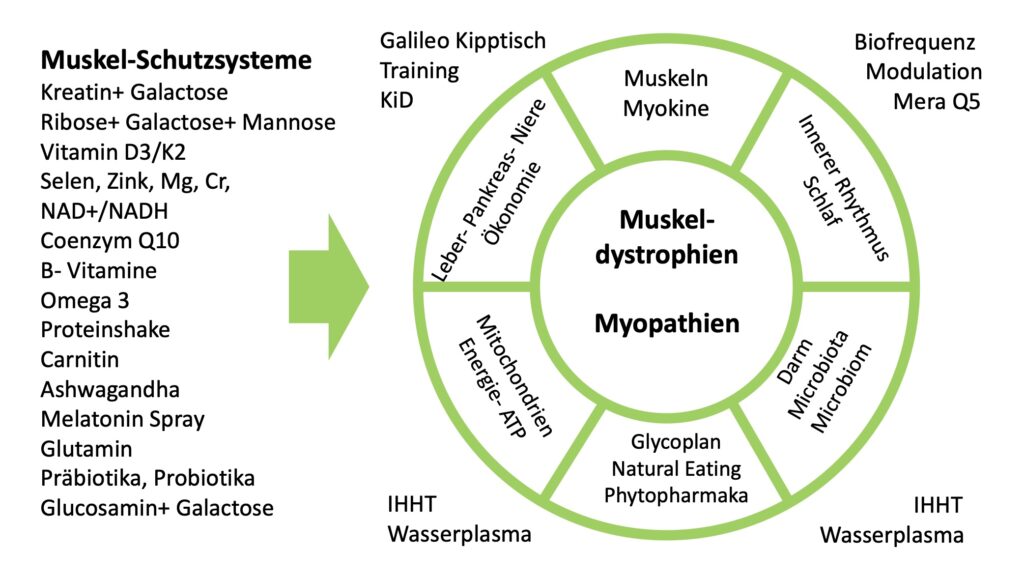
Es gibt verschiedene Myopathien. Es gibt angeborene Myopathien. Es gibt Myotonien, die in erster Linie mit der Spannung der Muskulatur im Zusammenhang stehen. Es gibt Myopathien, die von den Mitochondrien ausgehen.
Im Vordergrund steht die Muskelschwäche, dann aber auch der Muskelschwund. Diese können sich, nachdem viele Betroffene ein normales Leben geführt haben, „plötzlich“ und dramatisch sich verschlechtern – über Medikamente, über Infekte, über Entzündungen, über Alkohol und über Süßgetränke.
Es können zusätzlich noch Schluck-, Hör- und Herzmuskel Probleme entstehen. Einige Myopathien sind im Zusammenhang zu sehen mit einem dysregulierten Darm, sodass eigentlich toxische Metabolite aus dem Darm das Herz oder die Muskeln belasten können. Ebenso kann eine Schilddrüsen Erkrankung mit eine Rolle spielen.
Und sehr häufig sind Unterversorgungen (Mineralstoffe, Vitamine, Omega-3 und Aminosäuren) von Bedeutung.
Einige Myopathien gehen direkt in Entzündung im Körper, in die Faszien und die Muskeln über. Dann spricht man von „Autoimmunerkrankungen“. Diese fallen aber nicht aus dem heiterem Himmel, sondern sie haben eben eine konkrete Vorgeschichte!
Besonders hervorzuheben sind Myopathien, die durch Pestizide, Schwermetalle, Medikamente, Süßgetränke und Alkohol hervor-gerufen werden. Damit zeigt sich schon, ja, die körpereigenen Entgiftungssysteme werden überfordert und es muss behandelt werden.
Häufig findet man auch Intoleranzen gegen Nachtschatten-gewächse und Gluten. Also: immer glutenfrei und kuhmilchfrei, kaseinfrei. Dafür gibt es dann Ziege, Schaf, Cashew, Mandelmilch und Hafermilch und Kokosmilch.
Mukopolysaccharidosen (MPS) sind Stoffwechselerkrankungen, bei den es um zelluläre Speicherkrankheiten geht. D.h. dass bestimmte Substanzen im Körper, die normal abgebaut werden, nicht richtig abgebaut werden.
Wenn die Zucker-Eiweiß-Zuckermoleküle, Glykosaminoglykane und die andere Proteoglykane nicht richtig über Enzyme abgebaut werden, führt das zu einer kognitiven Entwicklungsverzögerungen, aber auch zu Skelettfehlbildungen.
Es entwickeln sich Dysostosen, Kleinwuchs, Hernien, Herzklappen-fehler, Gelenk Kontrakturen und so auch psycho-somatische Veränderungen. Auch Visusverlust, Schwer-hörigkeit und ganz häufig tatsächlich auch kognitive Dysfunktion sind möglich
Es können unterschiedliche Enzyme betroffen sein, beim Morbus Hurler oder beim Pfaundler-Syndrom ist es die Iduronidase. Beim Hunter-Syndrom werden andere Glykosaminoglykane falsch abgebaut. Beim Morbus Morquito herrscht ein Mangel an N-Acetyl-Galaktosamin-6-Sulfat-Sulfatase (Typ A) bzw. ein Mangel an beta-Galaktosidase (Typ B)
Insgesamt kann man hier nicht von Heilung sprechen, aber man kann mit Enzymersatztherapie und mit Darm-Leber-Medikamenten kompensatorisch viel tun und symptomatisch abfedern.
Dr. Mosetter Prinzip –
Vesalius GmbH
Obere Laube 44
78462 Konstanz
Tel +49 7531 7141 – 0
info@mosetter.de
Kontaktformular







